

Alport syndrome is a rare genetic disease that damages the tiny blood vessels in the kidneys. It may also cause hearing loss and vision problems. Alport syndrome is caused by a mutation in three different genes. Each of these genes provides instructions for making a kind of protein called “type IV collagen”. This protein, type IV collagen, plays an important role in the kidneys, specifically in the structures called glomeruli. Glomeruli are clusters of specialized blood vessels that remove water and waste products from the blood and helps make urine. Mutations in these genes result in abnormalities of the type IV collagen in glomeruli, which prevents the kidneys from properly filtering and allows blood and protein to pass into the urine. Because the kidneys are not properly working, gradual scarring of the kidneys occurs, eventually leading to kidney failure in many people with Alport syndrome. Type IV collagen is also an important component in the function of the inner ear and helps to define the shape of the eye and color of the retina. Due to this genetic mutation, the ears and eyes of those diagnosed with Alport syndrome may also be affected.
Alport syndrome has 3 genetic variances of the disease:
- X-linked Alport syndrome (XLAS) - This is the most common type. The disease is more severe in males than in females.
- Autosomal recessive Alport syndrome (ARAS) - Males and females have equally severe disease.
- Autosomal dominant Alport syndrome (ADAS) - This is the rarest type. Males and females have equally severe disease.
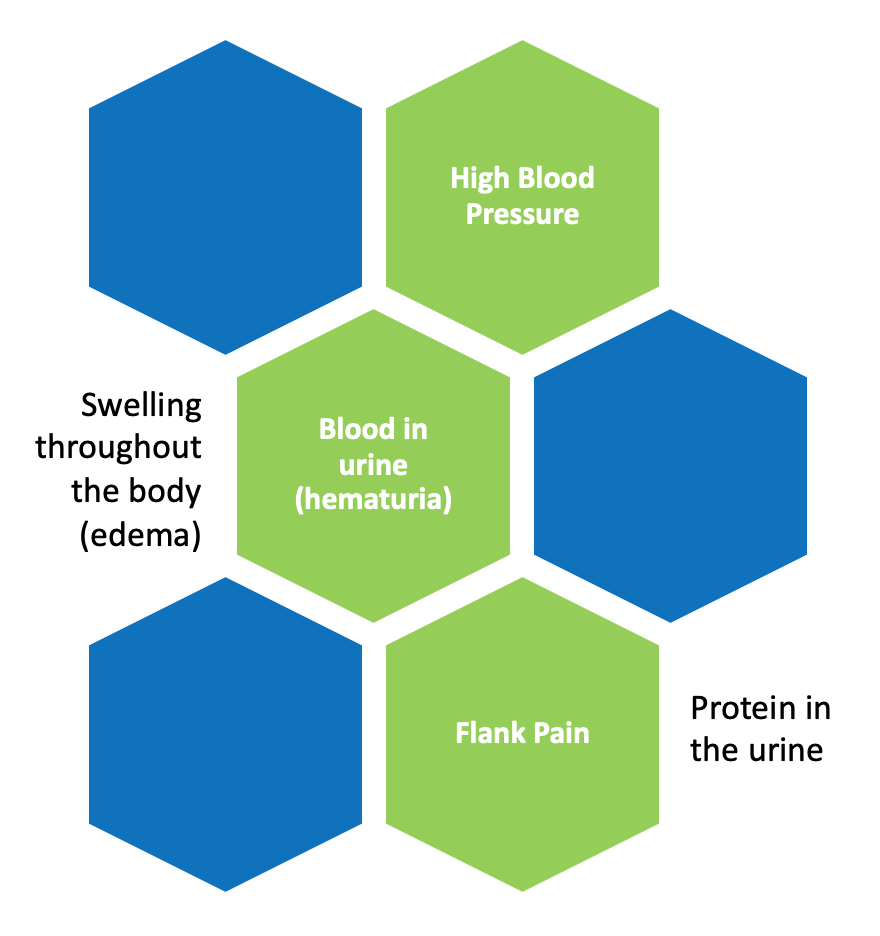
At first, there may be no symptoms, but often the first sign may be blood in the urine (hematuria). Over time, as the glomeruli become more and more damaged, kidney function is lost, waste products and fluids build up in the body, which can make you feel tired and sick. The condition can progress to end stage kidney disease (ESKD), which is when there is a total loss of kidney function. Dialysis or a kidney transplant will need to be considered if one develops ESKD.
Can Alport syndrome be prevented?
Alport syndrome is an inherited genetic condition, so it cannot be prevented. With early diagnosis, intervention and treatment, symptoms can be managed much better as medical advancements are being made.
How is Alport syndrome diagnosed?
The likelihood of diagnosis increases in individuals with a family history of Alport syndrome, kidney failure without known cause, early hearing loss or blood in the urine. For individuals with a family history of Alport syndrome, having the children in that family evaluated for the syndrome early in life is important to determine if they have the gene mutation. Often, the first symptom is blood in the urine, but this may not always be visible to the naked eye, which is why routine screening by a healthcare provider is important.
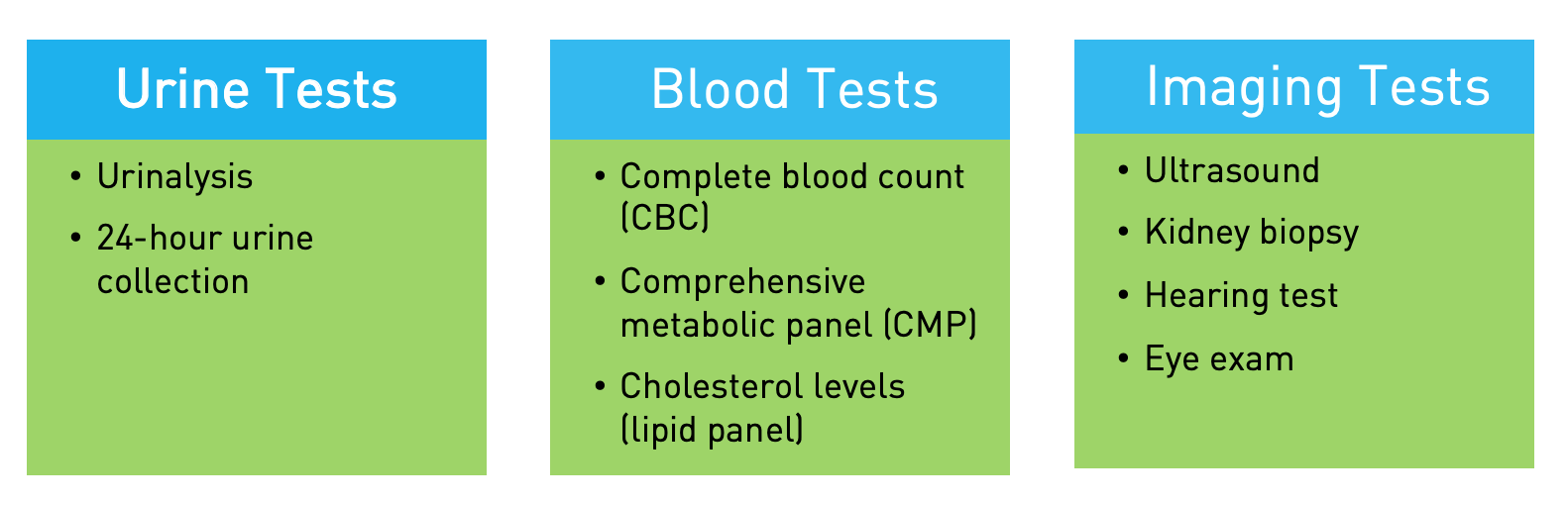
What tests can I expect that will help diagnosis if I have Alport syndrome?
What about genetic testing?
While a kidney biopsy is especially useful in the evaluation of people thought to have Alport syndrome, early genetic testing is becoming increasingly important. If a family history and clinical symptoms strongly suggest a diagnosis of Alport syndrome, genetic testing, can confirm the diagnosis, and provide useful information about how the disease will progress.
Genetic counselors are an important part of the genetic testing process. They help to identify families at possible risk of a genetic disorder, gather and analyze family history and inheritance patterns, calculate risks of recurrence, and provide information about genetic testing and related procedures. Genetic counselors work as members of a healthcare team, providing education and support to individuals and families at risk for, or diagnosed with a genetic condition and provide supportive counseling.
What is the treatment for Alport syndrome?
The goals of treatment include monitoring and controlling the disease and treating the symptoms. Your healthcare provider may recommend any of the following:

Although conservative medical treatment may slow the progression of kidney disease in Alport syndrome, there is no cure for the syndrome and currently no treatment has been shown to completely stop kidney decline caused by Alport syndrome. In many affected individuals, kidney function eventually worsens to the point where dialysis or a kidney transplant is required. Choosing the best treatment option requires a thorough discussion with all members of your healthcare team.
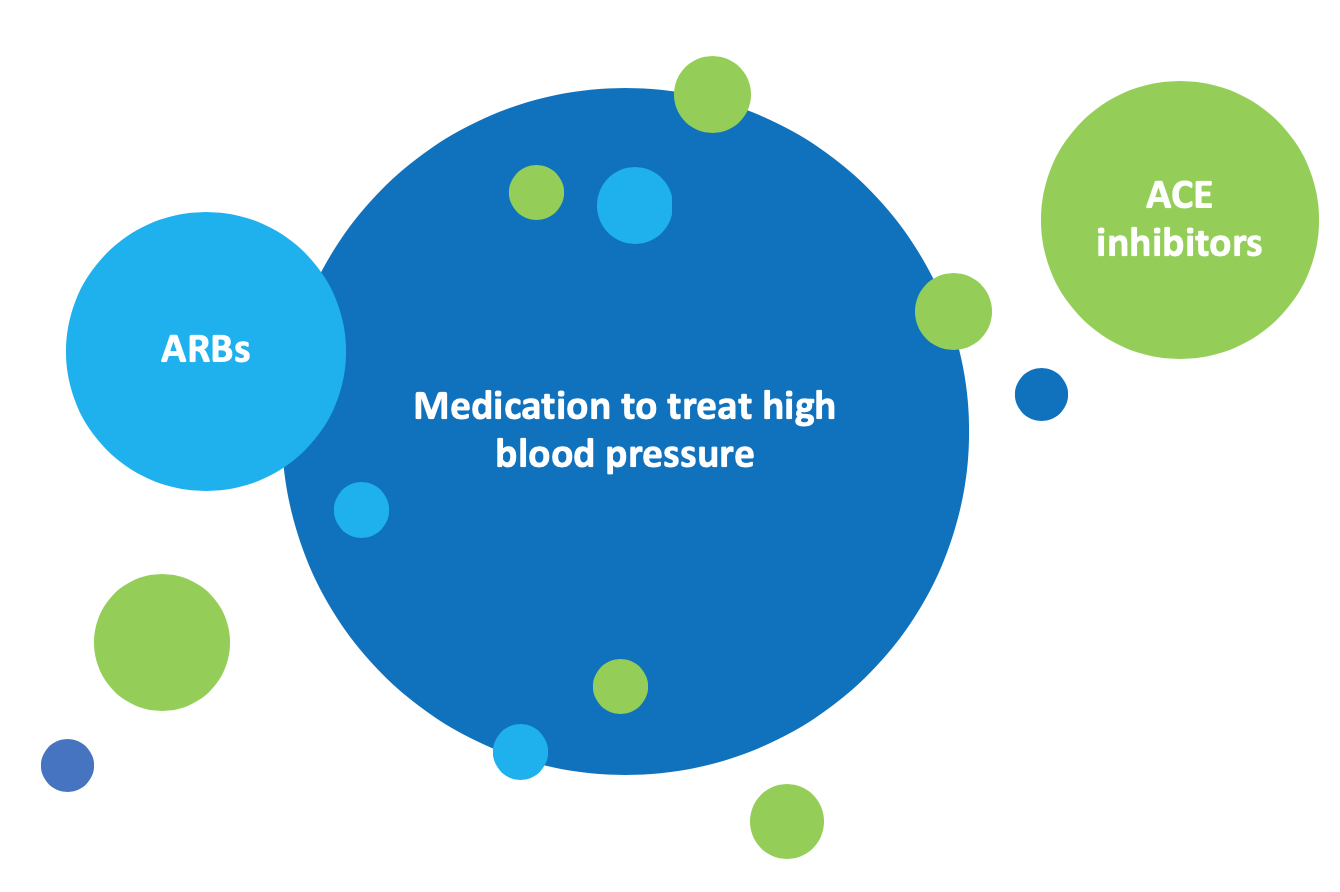
What specific medication(s) can I take to help with Alport syndrome?
Committing to taking the blood pressure medication as prescribed is essential. While blood pressure medications such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) help maintain a normal blood pressure, they also help prevent protein from spilling into the urine which is important in helping to slow down the loss of kidney function. Talk to your healthcare provider about which blood pressure medication will be best for you.
What about the complications of hearing and vision loss?
Individuals with Alport syndrome frequently develop hearing loss, caused by abnormalities of the collagen in the inner ear, which typically occurs during late childhood or early adolescence. Affected individuals may also have irregular lenses in the eye and an abnormal color of the retina (the light-sensitive tissue at the back of the eye). These eye abnormalities seldom lead to vision loss.
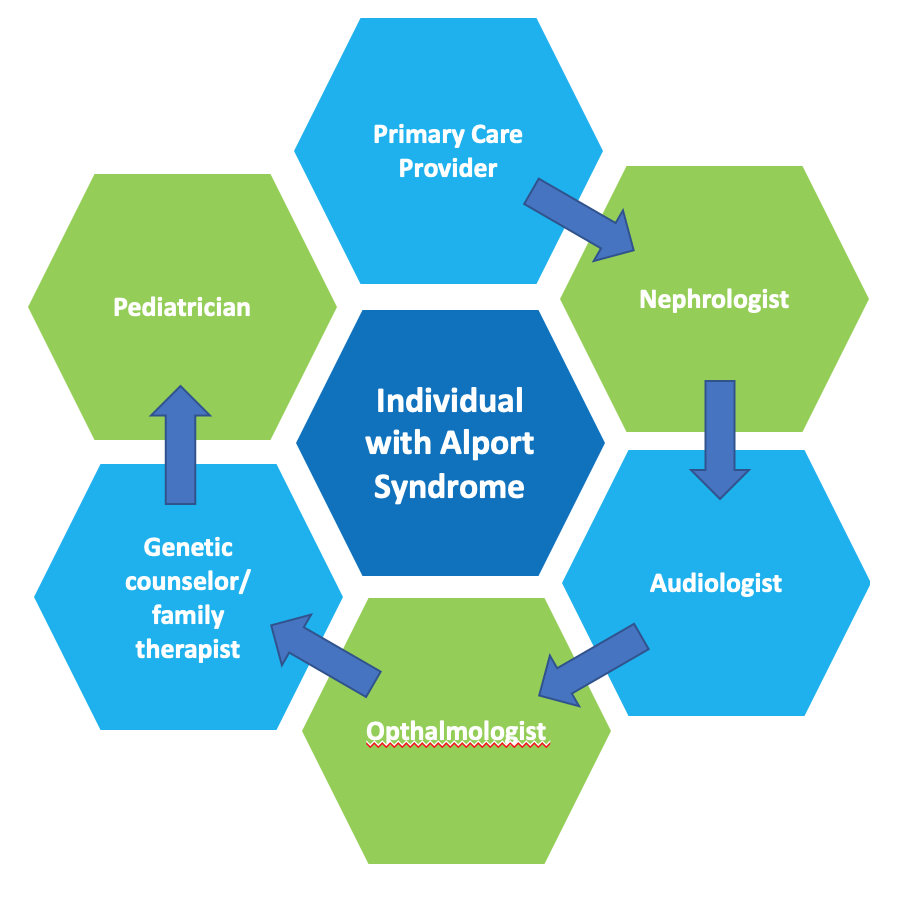
A comprehensive team of healthcare providers will be beneficial to support you and your family
Treatment is complex and will require coordination and collaboration among the whole healthcare team. The individual with Alport syndrome, alongside a primary care provider, nephrologist, audiologist, ophthalmologist, pediatrician (as applicable), and other team members must work together to ensure all components of the treatment plan are effective. Genetic counseling may also be recommended for affected individuals and their families, and a family therapist may also prove to be helpful as more than one family member may need treatment for Alport syndrome.
Additional Resources:
- Alport Syndrome Foundation
- AAKP Pocket Guide to Thinking about Genetics and Kidney Disease (Download)
- AAKP HealthLine Webinar: Understanding Genetic Conditions: What You Need to Know


